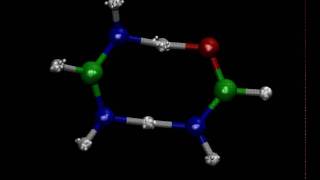
Media Summary: AIPIMD simulation of double proton transfer in Watson-Crick AT base-pair model FI-FA at 300 K. Sampling of phase-space is ... Hands-on Workshop Density-Functional Theory and Beyond: Accuracy, Efficiency and Reproducibility in Computational Materials ... Oliver Clarke (Columbia University) delivers a talk entitled 'High-resolution
Ab Initio Path Integral Molecular - Detailed Analysis & Overview
AIPIMD simulation of double proton transfer in Watson-Crick AT base-pair model FI-FA at 300 K. Sampling of phase-space is ... Hands-on Workshop Density-Functional Theory and Beyond: Accuracy, Efficiency and Reproducibility in Computational Materials ... Oliver Clarke (Columbia University) delivers a talk entitled 'High-resolution TYC Symposium: Physics and Chemistry of Electrified Interfaces, 20 May 2021: Water metal interfaces by Speaker: Michele CERIOTTI (EPFL, Lausanne, Switzerland) School in Computational Condensed Matter Physics: From Atomistic ...